
-
Publish Your Research/Review Articles in our High Quality Journal for just USD $99*+Taxes( *T&C Apply)
Offer Ends On
Publish Your Research/Review Articles in our High Quality Journal for just USD $99*+Taxes( *T&C Apply)
Offer Ends On
Armel Hervé Nwabo Kamdje* and Hervet Paulain Dongmo Fogang
Corresponding Author: Armel Hervé Nwabo Kamdje, Department of Physiology and Biochemistry, Faculty of Medicine, University of Garoua, P.O. Box 346, Garoua, Cameroon.
Received: August 03, 2024 ; Revised: August 26, 2024 ; Accepted: August 29, 2024 ; Available Online: September 06, 2024
Citation: Kamdje AHN & Fogang HPD. (2024) Role of Notch, Wnt and Hh Signaling in B-Cell Chronic Lymphocytic Leukemia inside the Bone Marrow Stromal Cells Microenvironment. J Can Sci Res Ther, 4(1): 1-9.
Copyrights: ©2024 Kamdje AHN & Fogang HPD. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Views & Citations
Likes & Shares
B-cell chronic lymphocytic leukemia (CLL) is a type of cancer of the blood and bone marrow and the most common form of leukemia in adults. It’s characterized by the accumulation of small, mature-appearing neoplastic lymphocytes in the blood, marrow, and secondary lymphoid tissues. The major signaling pathways involved in the tumorigenesis and in the resistance to the various anticancer therapies includes Notch, Hedgehog, and Wnt. Indeed, Notch signaling which regulates cell-fate determination during development and maintains adult tissue homeostasis was found recurrently mutated in CCL; about Wnt signaling, it also regulates crucial aspects of cell fate determination, cell migration, cell polarity, neural patterning and organogenesis during embryonic development and the Wnt/PCP pathway contributes to numerous aspects of CLL pathogenesis. Concerning Hedgehog, it exerts its biological effects through a signaling cascade that culminates in a change of balance between activator and repressor forms of glioma-associated oncogene (Gli) transcription factors and its inappropriate activation occurs in several human cancers, including hematological neoplasms.
Abbreviations: BFGF: Basic Fibroblast Growth Factor; BM: Bone Marrow; CD44: Cluster of Differentiation 44; CLL: Chronic Lymphocytic Leukemia; CXCR4: C-X-C Motif Receptor 4; FDC: Follicular Dendritic Cells; IRF4: Interferon Regulatory Factor 4; MSC: Mesenchymal Stem Cell; SDF-1: Stromal Cell-Derived Factor 1; TNF: Tumor Necrosis Factor; VEGF: Vascular Endothelial Growth Factor
INTRODUCTION
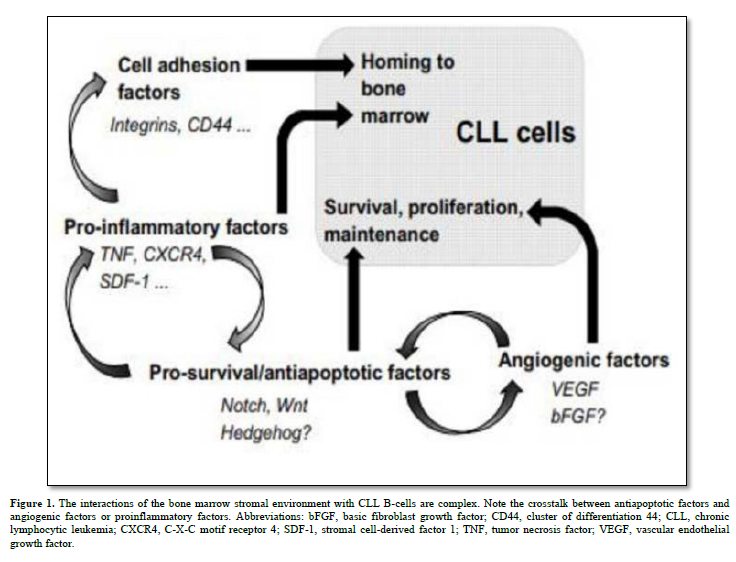
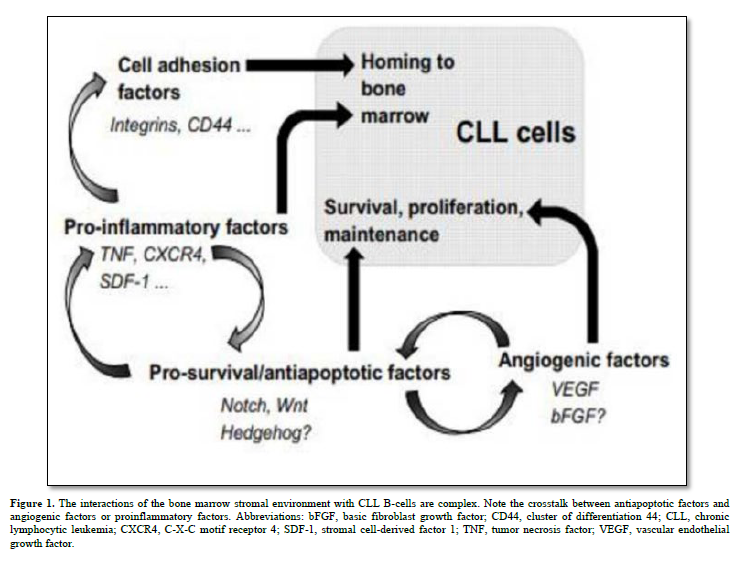
B-cell chronic lymphocytic leukemia (CLL) stands as the most common lymphoproliferative disorder characterized by increased production and accumulation of mature but dysfunctional B lymphocytes. CLL exhibits varying clinical courses, dictated by well-defined prognostic factors like CD38 and ZAP70 expression, V-gene mutation status, and specific gene profiles [1]. CLL manifests through low levels of immunoglobulins (IgM alone or with IgD), expression of B-cell associated antigens (CD19, CD20, CD21, CD23, and/or CD24) and expression of CD5 which is a T-cell associated antigen, in blood, secondary lymphoid tissues, and bone marrow (BM), closely associated with the stromal microenvironment. Malignant B-cells maintain responsiveness to diverse microenvironmental cues, offering therapeutic prospects. Notch, Wnt, and Hedgehog (Hh) pathways assume pivotal roles in the pathogenesis of various hematologic malignancies and solid tumors, governing essential cellular functions linked to tumorigenesis like proliferation, differentiation, and apoptosis [2] (Figure 1). This review summarizes and discusses recent findings on the involvement of Notch, Wnt, and Hh signaling pathways in CLL, their interactions with the stromal microenvironment and the use of these signaling pathways as target in the treatment of this hematopathology. It also considers new experimental evidence highlighting their roles in carcinogenesis.
NOTCH, Wnt AND Hh SIGNALING PATHWAYS AND STROMAL CELLS
Notch family
This family involves four receptors, Notch1-4 (Figure 1A), and five ligands: Jagged1, Jagged2, Delta-like (DLL) 1, 3, and 4 (Figure 1B). These are transmembrane proteins. When Notch ligands' extracellular domains bind to Notch receptors, it triggers conformational changes, revealing the S2 cleavage site. TNF-α converting enzyme then cleaves it (Figure 1C). Next, a second cleavage occurs at the S3 site in the receptor's transmembrane domain, mediated by the γ-secretase complex, releasing the Notch receptor intracellular domain (NICD), which moves to the nucleus.
In the nucleus, NICD forms a transcriptional complex (CBF1/RBP-Jk/Suppressor of Hairless/Lag1 or CSL), interacting with CBF1-dependent target genes. NICD also recruits co-activators like mastermind proteins (MAML1-3), bringing in the MED8-mediator complex, inducing downstream target gene transcription [3]. This cascade evolutionarily conserved, regulates the vascular development and physiology, multiple developmental processes of the Central Nervous System, is involved in the regulation of Nerve Stem Cells’ proliferation, survival, self-renewal and differentiation and has been associated with early neurodevelopment, learning and memory, as well as neurodegeneration [4].
Additionally, emerging data suggests crosstalk between Notch, Wnt, Hedgehog (Hh) pathways, and others like NF-κB, hypoxia, or TGF-β, expanding the range of target genes influenced by Notch, Wnt, and Hh systems [5].
Wnt family
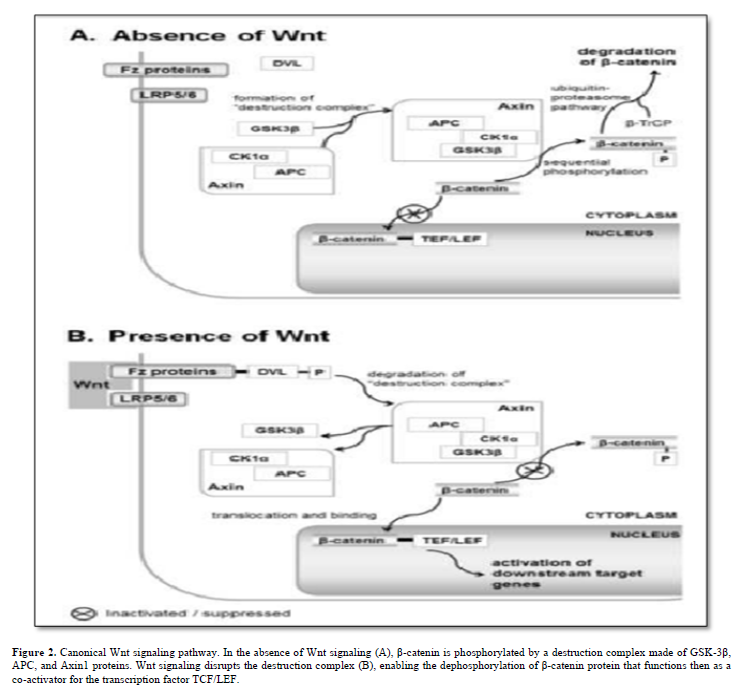
Wnt proteins, highly conserved signaling molecules, regulate cell-to-cell interactions in embryonic development and throughout an organism's life. They act by binding to the Frizzled (Fz)/low-density lipoprotein (LDL) receptor-related protein (LRP) complex on cell surfaces, initiating intracellular signaling cascades involving Dishevelled (Dsh), glycogen synthase kinase-3β (GSK-3), Axin, Adenomatous Polyposis Coli (APC), and β-catenin. Three main Wnt signaling pathways are recognized: (i) planar cell polarity, controlling cellular polarity and cytoskeletal interactions; (ii) Wnt/calcium, activating protein kinase C and calcium-calmodulin-dependent protein kinase II; and (iii) canonical Wnt/β-catenin [6].
The canonical Wnt/β-catenin pathway centers on β-catenin, encoded by the CTNNB1 gene. Without Wnt signaling, a destruction complex (GSK-3β, APC, and Axin1) phosphorylates β-catenin. Wnt activation disrupts this complex, allowing unphosphorylated β-catenin (at Ser 37 or Thr 41) to accumulate and co-activate the transcription factor TCF/LEF (T-cell factor/lymphoid enhancer factor). Figure 2 simplifies the current Wnt signal transduction model.
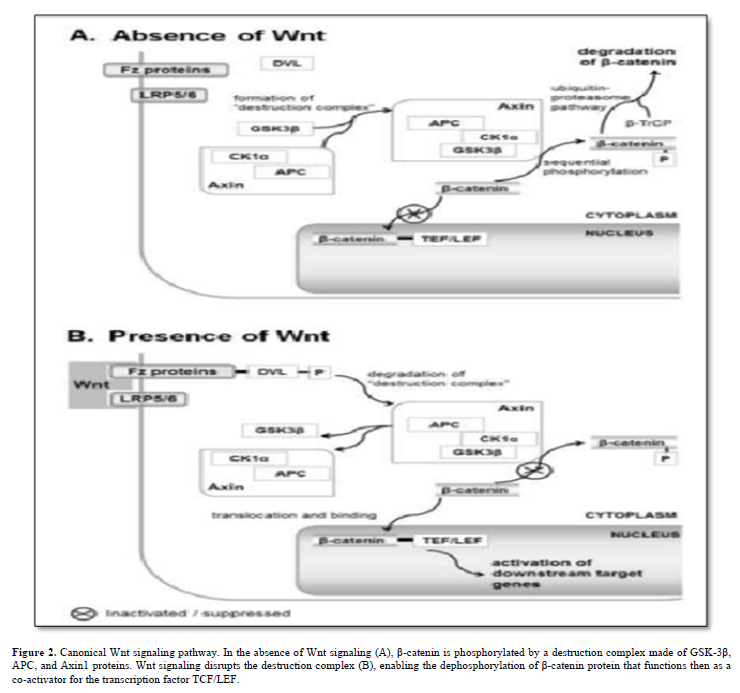
The TCF/LEF/β-catenin complex targets genes regulating the cell cycle and selectively governs Notch signaling pathway members, creating intricate cross-talk between Wnt and Notch signals [7].
Hh family
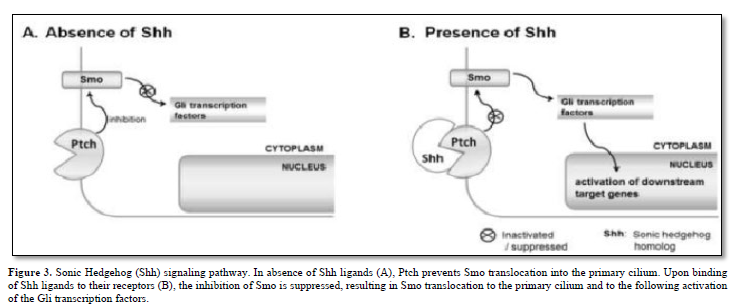
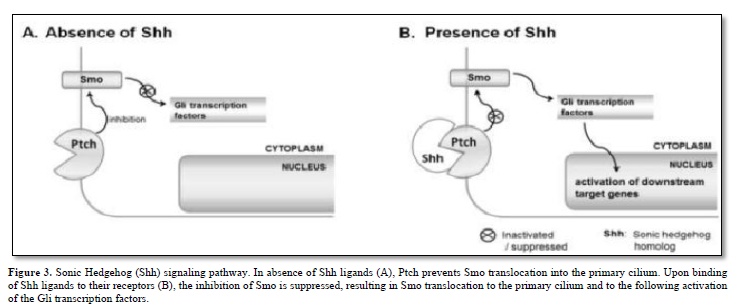
The Hh family of secreted signaling proteins plays crucial and evolutionarily preserved roles in embryo morphogenesis and cell cycle regulation. Within vertebrates, the Hh family comprises a minimum of three ligands: Desert Hh (Dhh), Indian Hh (Ihh), and Sonic Hh (Shh). Additionally, two homologs of the Drosophila transmembrane protein Patched (Ptch), namely Ptch1 and Ptch2, serve as Hh receptors and function as negative regulators of the signal transducer Smo. Furthermore, there are Gli zinc finger proteins, which are counterparts of the Drosophila Ci protein and function as transcription factors.
A prevailing hypothesis suggests that in the absence of Hh ligands, Ptch inhibits Smo translocation into the primary cilium by modulating the production and/or transport of small molecules such as oxysterols [8]. Typically, when Hh ligands bind to their receptors, this inhibition of Smo is alleviated. Subsequently, Smo relocates to the primary cilium and activates members of the Gli protein family as depicted in Figure 3.
BM stromal cells
Human bone marrow stromal cells are very rare skeletal stem cells and important constituents of the hematopoietic microenvironment. Recent discoveries have shed light on the origin of bone marrow (BM) stroma, revealing that it stems from adult multipotent non-hematopoietic stem cell precursors known as mesenchymal stromal cells (MSCs). These MSCs play pivotal roles within the BM hematopoietic niche by facilitating the maintenance and engraftment of hematopoietic stem cells. MSCs exhibit the remarkable abilities of self-renewal and differentiation into various mesodermal cell lineages, including fibroblasts, osteoblasts, adipocytes, and chondrocytes [9]. They display a consistent mesenchymal immunophenotype, characterized by the expression of markers such as CD105, CD44, CD73, CD90, and CD146. Furthermore, upon activation, both MSCs and their fibroblastic progeny exhibit potent and wide-ranging immune modulatory effects [10].
While originally identified in the bone marrow, where they serve as precursors for BM stromal cells and bone cells, MSCs are found in nearly every organ containing connective tissue, including lymphoid organs like the thymus and spleen.
NOTCH, Wnt AND Hh SIGNALING, STROMAL MICROENVIRONMENT, AND CLL
Notch signaling, stromal cells, and CLL B-cells
The Notch signaling pathway has been characterized as being deregulated in tumors, in particular in hematological malignancies. Indeed, Notch1 is one of the most commonly mutated genes detected in CLL patients. During CLL development, IRF4 has a crucial negative regulatory role in Notch signaling, thus lower IRF4 expression correlates with increased Notch2 level. As a consequence, Notch2 overexpression has been correlated with durable transcription of antiapoptotic stimuli such as Mcl-1. The interaction between CLL B-cells and BM stromal cells is thought to play a pivotal role in disease progression and therapy resistance due to stromal cells' ability to protect CLL B-cells from apoptosis induced by various treatments such as Fludarabine, Cyclophosphamide, Bendamustine, Prednisone, and Hydrocortisone [1,11].
Stromal cells and Notch molecules
Notch signaling, a critical pathway, has been found to be dynamic in human BM mesenchymal stem cells (MSCs) expressing Notch receptors and ligands like Jagged1, DLL3, and DLL4 [2]. The effects of γ-secretase inhibitor XII (GSI XII), a Notch inhibitor, on BM MSCs have shown that at higher concentrations (≥40.0 μM), these cells undergo apoptosis, indicating the specificity of Notch signaling effects [9]. Notch signaling has also been associated with osteogenic differentiation of MSCs and the regulation of hematopoietic stem cell niches [12]. However, the effects of Notch signaling vary in different stem cell populations.
In CLL patients, "blood-derived nurse-like cells" (bNLCs) have been isolated from peripheral blood mononuclear cells, which, in co-culture with CLL B-cells, enhance CLL B-cell survival through SDF-1/CXCR4 signaling. CLL B-cells closely associate with follicular dendritic cells (FDCs) in lymph nodes, where FDCs provide survival signals and upregulate the anti-apoptotic protein MCL-1 [13]. Altered regulation of Mcl-1 in CLL B-cells has been linked to chemotherapy resistance and prognosis [14].
These findings demonstrate that direct cell-cell contact between CLL B-cells and accessory cells such as FDCs or stromal cells creates a microenvironment that protects CLL B-cells from apoptosis.
Stromal microenvironment and CLL B-cells
BM stromal cells produce various cytokines, including Transforming Growth Factor (TGF)-β, colony-stimulating factors (CSFs), and interleukins (IL) 6, 7, and 10, influencing CLL B-cell behavior through these soluble factors and extracellular matrix proteins [15]. Notch1, Notch2, and Notch4 are implicated in regulating the expression of IL7 receptors on CLL B-cells. High levels of SDF-1/CXCL12, secreted by stromal cells, attract CLL B-cells via the CXCR4 chemokine receptor [16]. Stromal cells also impact CLL B-cell adhesion through integrins αLβ2 and α4β1.
Notch signaling and CLL B-cells
Notch signaling is believed to contribute to CLL B-cell survival and resistance to apoptosis.
Notch-1, -2 and -4 appeared to be mostly involved in both hBM-MSCs and hBM-MSCs-mediated CLL cell survival compared to low impact of Notch-3. All Notch receptors and ligands are constitutively expressed by CLL B-cells, with Notch2 involvement in CD23 overexpression. Inhibition of Notch signaling leads to apoptosis in CLL B-cells, while Notch stimulation enhances CLL B-cell survival through NF-κB activation, and expression of anti-apoptotic proteins such as c-IAP2 and XIAP. Recent study shows that Jagged-1, -2, DLL-3 ligands as well as Notch-1, -2 and -4 receptors expressed by both cell types resulted involved in CLL cell survival [17-23].
Additionally, the tumor suppressor protein p53 appears to interact with the Notch1 pathway, with p53-elevating agents inducing Notch1 expression [24]. Targeting Notch signaling with γ-secretase inhibitors or Notch2 down-regulation enhances the cytotoxic effects of p53-elevating agents in CLL B-cells [17].
Wnt signaling, stromal cells, and CLL B-cells
CLL B-cells exhibit high expression of Wnt family members such as Wnt3, Wnt14, and Wnt16, along with the receptor Frizzled3 [25]. Canonical Wnt signaling, involving β-catenin and transcription of target genes like c-Myc and Cyclin D1, plays a role in CLL B-cell survival [25]. Dysregulation of the Wnt antagonist Dickkopf1 (DKK-1) has also been reported in CLL [26]. Wnt/β-catenin signaling particularly plays a role in hematopoiesis and has been implicated in the control of proliferation, survival, and differentiation of hematopoietic cells [27]. Anomalous Wnt signaling has been associated with several cancers, the most prominent ones being colorectal, breast, lung, oral, cervical, and hematopoietic malignancies.
Interactions between CLL B-cells and stromal cells in pseudo follicular proliferation centers involve Wnt proteins, possibly contributing to CLL B-cell survival and proliferation [28]. Inhibition of Wnt signaling with Dickkopf1 (DKK-1) or soluble Frizzled-related protein 2 (sFRP-2) reduces CLL B-cell survival in co-culture with stromal cells [29].
Hh signaling, stromal cells, and CLL
Several previous works have mentioned the crucial role of tumor cell-microenvironment interactions in driving CLL proliferation and survival, contributing to its pathogenesis [30]. While the exact CLL origin remains debated, it's established that CLL B-cells consistently engage stromal cells in bone marrow and lymph nodes, supporting in vivo survival. Hh signaling's role in hematopoiesis, normal and malignant, is debated. Some report self-renewal defects [8], while others don't observe hematopoietic phenotypes, possibly due to timing differences [29]. Importantly, Hh ligands, especially Ihh, enhance CLL B-cell survival in the stromal microenvironment [29], and Ihh is highly expressed in hematopoietic cell lines [8]. Impaired Hh signaling in CLL B-cells may stem from increased stromal Hh ligands or intrinsic pathway defects. Overexpression of Gli1, Gli2, and negative regulator Sufu correlates with CLL progression and poor clinical outcomes [29]. Selective Gli inhibition reduces CLL B-cell survival and induces apoptosis co-cultured with BM stromal cells and Fludarabine. Recent study shows that a large proportion of patients have CLL cells with activated Hh signaling, which is associated with early disease progression and enhanced sensitivity to inhibition of GLI1 [31]. Pharmacological Hh inhibition doesn't affect normal hematopoiesis, highlighting specific CLL Hh signaling alterations and the potential of Gli targeting to enhance chemotherapy susceptibility.
Targeting Gli1 pharmacologically to inhibit Hh signaling is a promising CLL therapy approach. Cyclopamine inhibitor studies effectively suppressed stromal-induced CLL B-cell survival [32]. However, even without the inhibitor, CLL B-cell survival with stromal cells exceeded Shh-enriched medium. This underlines Hh signaling's centrality but not its exclusivity in enhancing CLL B-cell survival.
Stromal-released Hh ligands also support cancer cell survival in other types, contributing to increased proliferation and decreased apoptosis, akin to B-cell malignancies [29]. Gli1 and Gli2 among the Gli transcription factors are extensively studied in tumor development [29]. Hh ligands from stromal cells promoting malignant cell survival are identified in non-Hodgkin lymphoma and multiple myeloma [8]. Recent studies have shown that Notch, Wnt and Hh signaling pathways may not only be targets for the treatment of CLL, but also help understand their role in providing resistance to various cancer treatment modalities [33].
THERAPEUTIC TARGETING OF NOTCH, Wnt, AND Hh SIGNALING IN CLL
Targeting Notch Signaling in CLL
Diverse studies identified recurrent mutations in hematological malignancies making Notch one of the most desirable targets in leukemia. The Notch signaling mediates resistance to therapy and controls cancer stem cells supporting the development of on-target therapeutic strategies to improve patients’ outcome. Several classes of drugs with different targets have already been published. Abs directed to the EGF-repeat region (WC613) and to the NRR NOTCH1 domain (WC75) inhibited canonical Notch signaling in vitro by repressing Notch transcriptional targets such as Hes1 and DTX1 genes. Similarly, OMP-52M51 efficiently blocked canonical Notch signal and reduced Notch activation. Inhibitors of the γ-secretase complex (GSIs) like MK-0752, that target all NOTCH receptors, are purposed in cancers where NOTCH1 mutations are common (T-ALL, CLL) [34]. PF-03084014 induces selective apoptosis in primary CLL cells carrying NOTCH1 mutations and synergize with fludarabine in a stroma coculture model system [35]. In CLL bepridil exerted an anti-leukemia activity associated with Notch1 inhibition [34].
Although several other studies exist, these few examples demonstrate that the Notch signaling pathway represents a significant avenue in the treatment of CLL.
Targeting Wnt Signaling in CLL
The dysregulation of Wnt signaling in CLL B-cells has led to investigations into therapeutic strategies targeting this pathway. Several approaches have been explored to modulate Wnt signaling in CLL.
Dickkopf1 (DKK-1) is a secreted antagonist of Wnt signaling that inhibits the binding of Wnt ligands to their receptors [36]. CLL B-cells secrete DKK-1, and its expression is associated with adverse clinical outcomes [37]. Neutralizing DKK-1 with monoclonal antibodies or small molecules could potentially disrupt the pro-survival effects of Wnt signaling in CLL. Another approach involves the use of soluble Frizzled-related proteins (sFRPs), which can sequester Wnt ligands and prevent their interaction with Frizzled receptors [38]. In CLL, sFRP-2 is expressed by stromal cells and may contribute to CLL B-cell survival in the microenvironment [29]. Targeting sFRP-2 or other sFRPs could be a viable therapeutic strategy. Furthermore, small molecules that inhibit specific components of the Wnt pathway, such as β-catenin or TCF/LEF transcription factors, are being investigated for their potential to disrupt Wnt signaling in CLL B-cells.
Among used molecules, we can list fludarabine, cyclophosphamide, rituximab and, more recently, also novel inhibitors that target pro‐survival B‐cell receptor or anti‐apoptotic B‐cell lymphoma 2 (BCL2) signaling (ibrutinib, idelalisib, venetoclax, obinutuzumab and ofatumumab) [39].
Targeting Hedgehog (Hh) Signaling in CLL
Hedgehog (Hh) signaling has emerged as a potential therapeutic target in CLL due to its role in promoting CLL B-cell survival and interaction with the stromal microenvironment. Inhibition of Hh signaling has been explored using Smoothened (SMO) antagonists, such as LDE225. However, SMO inhibitors would be able to block ligand-dependent Hh pathway activation, but they would still be ineffective in CLL cells that harbor GLI1 proactivating mutations of SMO or downstream molecules. Another observation is that viability of GLI1+ CLL cells, but not GLI1-CLL cells, is significantly reduced by GLI1-specific small interfering RNA (siRNA) or by the GANT61 small molecule inhibitor of GLI1.
Therapists are attracted by inhibitors such as GANT61 because they may successfully operate in all the GLI1+ CLL cells by blocking either ligand-dependent or ligand-independent Hh pathways [40].
Notably, the crosstalk between Hh and Notch signaling pathways suggests that targeting both pathways simultaneously may be more effective in overcoming CLL resistance mechanisms. Combining Hh inhibitors with other targeted agents or chemotherapy regimens warrants further investigation.
NOTCH, Wnt AND Hh DEREGULATION IN CLL
The Notch signaling pathway has recently emerged as a contributor to CLL B-cell survival and resistance to apoptosis [17]. Di Ianni and colleagues delved into the mechanisms behind this in 133 consecutive CLL cases, examining Notch1 PEST domain mutations. They also screened for Notch2 mutations in 73 CLL cases due to structural similarities between Notch1 and Notch2. Surprisingly, only a minority (7/133, 5.3%) displayed Notch1 PEST domain mutations, and no Notch2 mutations were found, ruling out its involvement in altered Notch signaling in CLL. Despite their rarity, Notch1 PEST domain mutations were found to activate Notch signaling in CLL, similar to their association with poor prognosis in some T-cell acute lymphoblastic leukemia patients [41].
Recent studies have noted an increase in cases with Notch1 activation from diagnosis (8.3%) to Richter's transformation (31.0%) and in chemo refractory CLL (20.8%). This emphasizes the independent predictive value of Notch1 activation for poor survival, particularly in clinically aggressive disease forms [33]. Other researchers have made similar observations, solidifying Notch1 mutations as independent predictors of CLL B-cell survival. Studying CLL B-cells with Notch1 mutations in direct contact with bone marrow stromal cells, comparing them to CLL B-cells without such mutations, could provide valuable insights.
In contrast, constitutive activation of the Hh pathway through Ptch and Smo gene mutations is associated with various solid tumors but does not appear to play a significant role in CLL B-cells. Instead, Gli1, rather than Hh ligands or Smo, seems to autonomously promote cell survival in CLL B-cells [40].
Wnt signaling gene overexpression and reactivation have been reported in CLL, potentially contributing to the apoptosis defects characteristic of this malignancy. However, the rarity of β-catenin amino-terminal mutations in affected cells suggests that mutations may not be the primary underlying cause of Wnt signaling activation.
Altogether, the CLL B-cell coding genome is complex, with Notch, Wnt, and Hh signaling deregulations playing crucial roles. Managing these aberrations holds promise for both prognostic and therapeutic implications.
REGULATION OF OTHER SIGNALING PATHWAYS BY NOTCH, Wnt AND Hh SIGNALING
The Notch, Wnt, and Hh signaling pathways exert direct impacts on cell survival while also influencing other pathways indirectly. For instance, heightened c-MYC, a cell cycle-regulating transcription factor, has been associated with leukemia cell resistance to apoptosis. Notably, Notch signaling activation elevates c-MYC levels in leukemia cells [42]. Similarly, activation of the protein kinase Akt, an apoptosis inhibitor, follows Notch signaling activation, possibly due to the suppression of the negative regulator of phosphatidylinositol 3-kinase, Pten [43].
In CLL patient B-cells, Notch1, Notch2, and Notch4 have been observed to modulate cytokine expression, affecting pathways such as NF-κB, Akt/PI3K, and JNK/STAT, linked to apoptosis resistance. For instance, CLL B-cells show constitutive phosphorylation of STAT1 and STAT3 serine residues, unlike healthy B-cells [44].
The role of the Hh pathway in leukemia, lymphoma, and multiple myeloma is debated. In multiple myeloma, Hh pathway activation relates to undifferentiated stem cell expansion via a paracrine mechanism. In lymphoma, stromal cells produce Hh ligands supporting cell survival. In diffuse large B-cell lymphoma, autocrine Hh activation causes cell-cycle arrest and apoptosis. A similar autocrine activation is suggested in B-cell acute lymphoblastic leukemia [45].
LEF-1, a prosurvival factor in CLL, is expressed in monoclonal B-cell lymphocytosis. Notably, Notch and VEGF pathways interact with Wnt through regulation and physical interactions with pathway members active in CLL. Future research should explore how CLL Wnt activity interacts with other oncogenic pathways. Furthermore, apart from its Wnt signaling role, LEF-1 acts independently of β-catenin, functioning as a context-dependent transcription factor and interacting with intracellular Notch [46]. These functions require further investigation in future studies.
CONCLUDING REMARKS
It has become evident today that Notch, Wnt, and Hh signaling play vital roles in regulating cell fate and differentiation during normal and leukemic hemato-lymphopoiesis development. Notably, the oncogenic potential of Notch signaling in T-cell acute lymphoblastic leukemia was discovered. Interest in this pathway, along with Wnt and Hh signaling in CLL research, surged after identifying prevalent genetic alterations like Notch1 gain-of-function and gene mutations in T-cell acute lymphoblastic leukemia cells. Recent studies have confirmed these roles in CLL pathogenesis. Yet, the intricate interplay among these pathways and others involved in cell survival and proliferation complicates pinpointing their exact functions in normal and neoplastic lymphoid cell differentiation.
Nevertheless, these findings reshape our understanding of stromal support in CLL. Understanding the anti-apoptotic effects of Notch, Wnt, and Hh signaling has led to novel leukemia therapies. Future research may uncover more pathways contributing to CLL B-cell resistance to apoptosis, potentially introducing new anti-cancer drug classes.

No Files Found




Internationally Accepted
Share Your Publication :