
-
Publish Your Research/Review Articles in our High Quality Journal for just USD $99*+Taxes( *T&C Apply)
Offer Ends On
Publish Your Research/Review Articles in our High Quality Journal for just USD $99*+Taxes( *T&C Apply)
Offer Ends On
Anne Hucks Moxley and David Reisman*
Corresponding Author: David Reisman, Department of Biological Sciences University of South Carolina, Columbia, SC 29208 USA
Received: June 08, 2022 ; Revised: July 20, 2022 ; Accepted: July 23, 2022 ; Available Online: August 10, 2022
Citation: Moxley AH & Reisman D. (2022) The WRAP53 Gene is induced in Response to DNA Damage: A proposed Role for the p53 Tumor Suppressor. J Can Sci Res Ther, 2(1): 1-5.
Copyrights: ©2022 Moxley AH & Reisman D. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Views & Citations
Likes & Shares
Abstract
The Wrap53 mRNA transcript regulates expression of the p53 tumor suppressor gene by interacting with the 5'-untranslated region of the p53 mRNA transcript. The interaction of p53 mRNA and Wrap53 mRNA, enhances stability of the p53 mRNA and may enhance translation, thus increasing the levels of active p53 protein in the cell. This allows the cell to respond to DNA damage through p53-mediated cell cycle arrest or apoptosis and defects in this pathway lead to tumor formation. A previous analysis determined that the Wrap53 gene is regulated at the transcriptional level in response to DNA damage. A transcription factor database search revealed p53 as a potential protein that could bind to numerous sites in the WRAP promoter and studies investigating these possible interactions indicated that there is a clear transcriptional response to treatment of cells with agents that damage DNA and that p53 protein can bind to at least two of the identified sites in the WRAP promoter. Additional evidence points to the distinct possibility that p53 may be participating in activation of the WRAP gene in response to DNA damage, thus acting as part of a positive feedback loop.
Defects in this pathway will disrupt the normal DNA damage response ultimately leading to tumor formation.
Keywords: WRAP53 p53, Transcription regulation, DNA damage, Potential positive feedback loop
INTRODUCTION
The p53 tumor suppressor gene is one of the most commonly mutated genes identified in a myriad of human cancers [1,2]. The p53 protein is capable of inducing a cell-cycle checkpoint that results in either cell-cycle arrest or apoptosis in response to DNA damaging agents or cell stressors [3-7]. A number of years ago, a gene oriented in the opposite direction relative to p53 and overlapping the p53 first exon, was identified by Mahmoudi [8] and designated Wrap53 (NCBI Gene ID: 55135; ref. 9).
The study by Mahmoudi [9] determined that of the numerous alternatively spliced products (15 in all), one transcript, designated Wrap53 was shown to regulate p53 levels by binding to the 5' untranslated region of the p53 mRNA transcript [8]. In cooperation with additional proteins, notably the protein CTCF [10], the induced expression [11] and binding of Wrap53 mRNA induces stability and translation of the p53 mRNA, which increases the levels of active p53 protein in the cell. This ultimately allows the cell to respond to DNA damage via p53-mediated cell cycle arrest or apoptosis. Therefore, the increase in the expression of Wrap53 mRNA is able to induce p53-dependent apoptosis at least in cisplatin-treated human U2OS cells [11]. However, generality of this response and the mechanism of induction of the Wrap53 gene in response to DNA damage remained an open question.
Further studies have now found that the Wrap53 gene is regulated at the transcriptional level and in searching for DNA-binding transcription factors that may be responsible for this crucial induction of WRAP53α, it was observed that the p53 protein itself binds to a specific site or sites in the WRAP53α promoter and thus appears to participate in inducing its expression in response to DNA damage as part of an apparent positive feedback loop.
INDUCTION OF WRAP53 RNA AND p53 PROTEIN AFTER TREATMENT OF CELLS WITH DNA DAMAGING AGENTS
Findings reported by Yuan [11] demonstrated that there was a 3- to 40-fold increase in the level of Wrap53 mRNA after treatment of U2OS cells with 5 to 20 M cisplatin. In order to confirm and extend these findings, additional cells types such as U2OS, HCT116, and MCF7, all of which express wild-type p53 were treated a with a variety of DNA damaging agents. Gene expression analysis by qPCR demonstrated an increase in the level of WRAP53 product after treatment of U2OS and HT116, whereas MCF7 cells resulted in a very moderate increase in WRAP53 levels indicating that different regulatory mechanisms may be a play in different cell type. This is currently being further explored.
THE WRAP53 PROMOTER IS INDUCED IN RESPONSE TO DNA DAMAGE
The WRAP53 gene is induced at the transcriptional level in response to DNA damage. This was demonstrated by evaluating the activity the WRAP53 promoter linked to a luciferase reporter vector as well as by measuring the levels of WRAP53 mRNA by qPCR. Transcriptional regulation of the WRAP53 promoter was shown to be modulated in its expression after treatment of cells with agents that damage DNA. In fact, all the DNA damaging agents used induced the WRAP promoter, but to varying degrees. Treatment with actinomycin D and cisplatinum led to an approximately 2.5-fold elevation in promoter activity, camptothecin to a 3-fold elevation, and doxorubicin and etoposide to a 6.5- and 4.5- fold elevation, respectively. Therefore, although the WRAP53 promoter is induced in response to DNA damage or induced cellular stress, the extent of the response may depend on the type of DNA damage or the type of drug used. That a drug like cisplatin induced an 8-fold elevation in the level of WRAP53 RNA yet only a 2-fold elevation in promoter activity suggests that other regulatory mechanisms may also be at play, suggested by the lack of robust expression in MCF7 cells. One possibility is that regulation occurs post- transcriptionally through altering the extent of WRAP mRNA stability. Overall, these findings are consistent, as described below, and with published large-scale analyses of publicly available p53 transcriptional networks, with wild-type p53 contributing to WRAP53 expression [12,13].
p53 AS A DNA-BINDING TRANSCRIPTION FACTOR OF THE WRAP53Α PROMOTER
With these findings that WRAP53 is induced in response to DNA damage, the question remained: which transcription factors could be responsible for upregulating WRAP53α in response to DNA damage? Bioinformatic recognition algorithms (PROMO; ref. 14) were employed to detect transcription factors that could, in theory, bind to the promoter. Sixty-six possible transcription factors, including p53, were identified by the software as potentially being able to bind to the WRAP53α promoter/first exon sequence [14]. However, due to the obvious functional and physical relationship between p53 and WRAP53α [8], a potential p53/WRAP53α interaction as a mode of transcriptional regulation seemed viable.
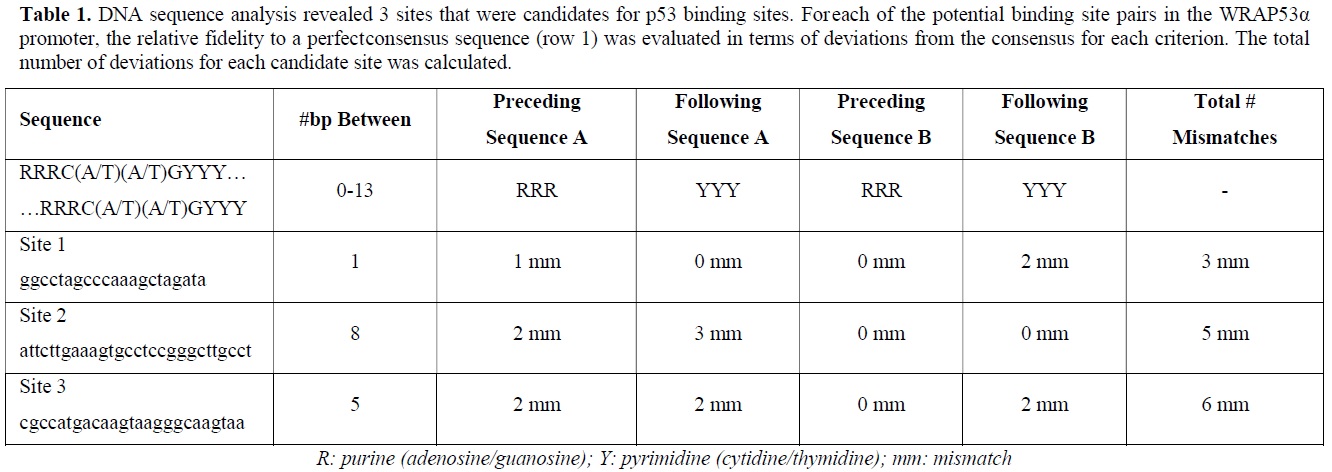
According to the p53 literature, p53 binds as a tetramer to two half-sites separated by 0-13 bp [15]. A perfect consensus sequence is considered to consist of two half-sites, A and B, each bearing the sequence C(A/T)(A/T)G, separated by 0-13 bp.
Furthermore, each half-site is preceded by three purines and followed by three pyrimidines [15,16]. Therefore a canonical p53 consensus binding site would be: RRRC(A/T)(A/T)GYYY--(0-13bp)-RRRC(A/T)(A/T)GYYY. By these criteria three putative p53 binding sites, designated sites 1,2 and 3, have been investigated (Table 1).
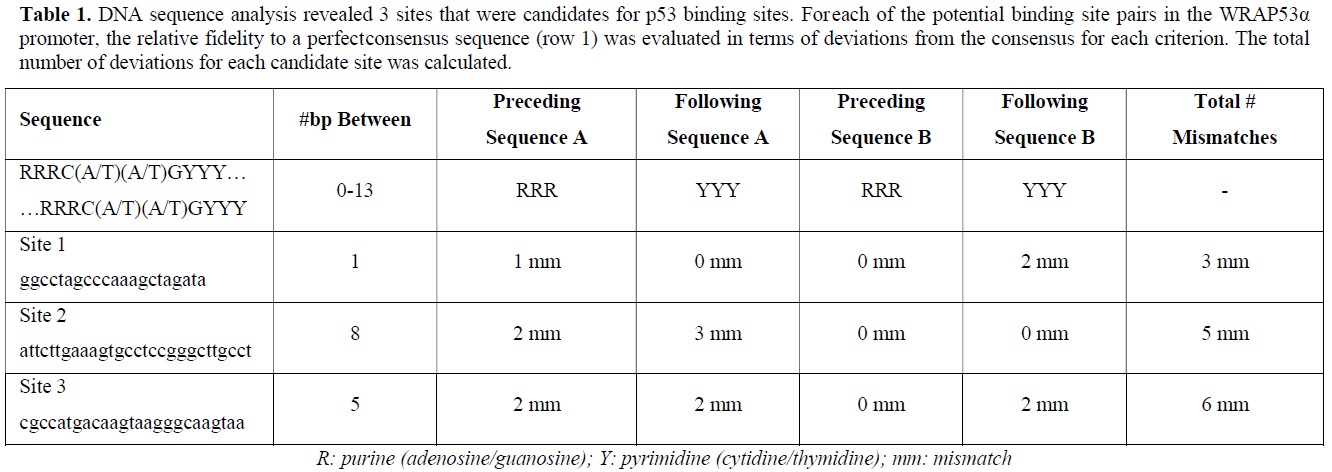
ChIP ANALYSIS DEMONSTRATES BINDING OF p53 TO THE WRAP PROMOTER
ChIP analysis is extremely useful for identifying factors that bind to specific DNA sequences in vivo. By employing ChIP it was found that in response to treatment with 1 μM doxorubicin, p53 binds to site 1 with an intensity that was approx. 3-fold greater than a negative control; this was consistent with a p53/p21 interaction that was also approx. 3-fold greater than the negative control. However, p53 interactions at sites 2 and 3 were deemed unlikely or at best, very weak, yielding 1.5- and 1.7-fold greater intensity than background, respectively. In response to treatment with 35 μM cisplatin however, p53 appeared to bind to sites 1, 2, and 3 with intensities that were 2.5-, 10-, and 3-fold greater than the negative control, respectively, while p53 bound to p21 with an intensity that was 3-fold greater than background. Therefore, putting all these findings together, p53 appears to bind to the WRAP53α promoter at the candidate p53 binding sites, and that the degree and location of binding may be dependent on the type of DNA damage. These data are also consistent with publicly available p53-binding databases that indicate that p53 binds to the WRAP promoter [12,13].
p53 BINDING SITES ARE REQUIRED FOR A COMPLETE DNA-DAMAGE INDUCTION OF THE WRAP PROMOTER
In order to determine whether one or more of the identified p53 binding sites are required for the induced expression of WRAP53, three mutant WRAP53α promoters linked to the luciferase gene were created, each lacking putative p53 binding sites 1,2, and 3. Transfection of these reporters constructs into U2OS cells and treatment with 1 M doxorubicin for an additional 24 or 48 h revealed that the site 1 mutant showed a moderate reduction in activity, yet no apparent reduction in activation from the site 2 or site 3 mutants. Since upon treatment of cells with doxorubicin, p53 was found to bind to sites 1 and 2 but not significantly if at all to site 3, a 4th mutant lacking both sites 1 and 2 led to the elimination of induced activity after treatment with doxorubicin.
Therefore, while the loss of each putative p53 binding site on its own may not be sufficient to lose promoter activity, the loss of sites 1 and 2, both found to bind p53, leads to loss of induction. Therefore, putting all the data together, one conclusion is that the binding of p53 to a pair of target sites on the WRAP promoter is required for WRAP promoter induction upon DNA damage.
CONCLUSION
p53 was first reported to be regulated post-transcriptionally by the mRNA transcript of its antisense gene, WRAP53α, in 2009 [8]. When siRNA-mediated knock-down of the WRAP53α transcript occurred, the p53 transcript was degraded rapidly and levels of translation were not sufficient to elicit a tumor suppressive effect [8]. More recent finding has provided insight into the behavior of the WRAP53α promoter, particularly its response to DNA damage and its interaction with p53 protein. While it had been clearly established that WRAP53α is induced in response to DNA damage [11], more recent studies have extended these results to show that it is transcriptional upregulation that ultimately leads to this induction; furthermore, the p53 protein, as a transcription factor, binds to the WRAP53α promoter at several different locations, leading to increased mRNA production in response to DNA damage.
Subsequently, through a bioinformatics search, a number of potential p53 binding sites in the WRAP promoter were identified and assayed to determine the possibility of p53- mediated transcriptional regulation of WRAP53.
ChIP assay allow the identification of protein-DNA interactions that could potentially occur in vivo and accordingly, doxorubicin, a topoisomerase II inhibitor, promotes binding of p53 to site 1, while cisplatin, a cross-linking agent, appears to promote binding to sites 1, 2, and 3. Ongoing studies will determine whether distinct post-translational modifications of p53 lead to differential DNA binding specificity. Consistent with this notion, activated p53 target genes when compared between cisplatin- and adriamycin-treated cells, found that that different drugs induce different classes of genes in an isoform-specific way [17]. Although a number of published ChIP assays looking at genome-wide p53 binding have indicated that p53 can bind to the WRAP promoter in multiple cell types, the significance of this interaction was not clear [12,13].
Does a positive feedback loop exist between p53 and WRAP53α?
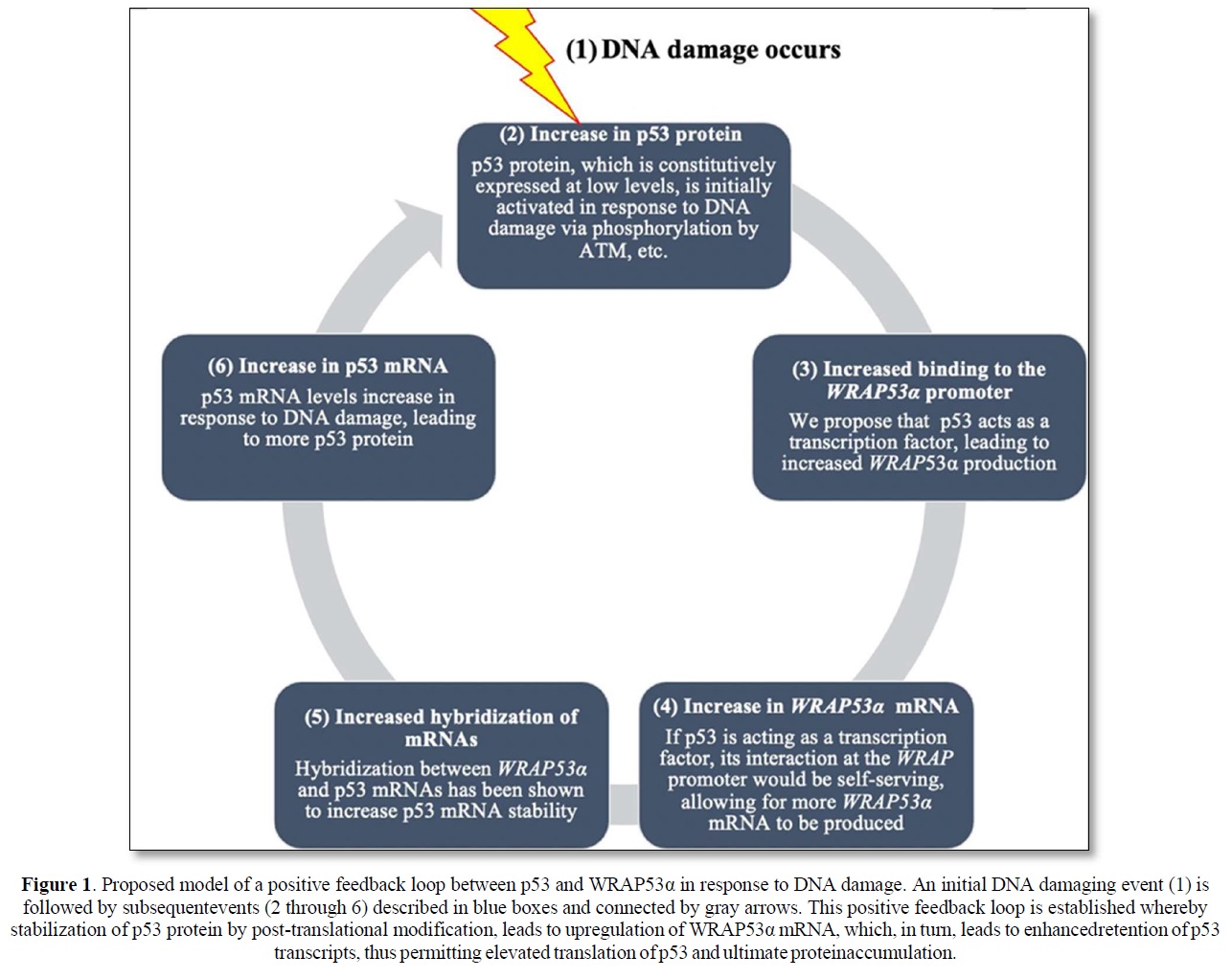
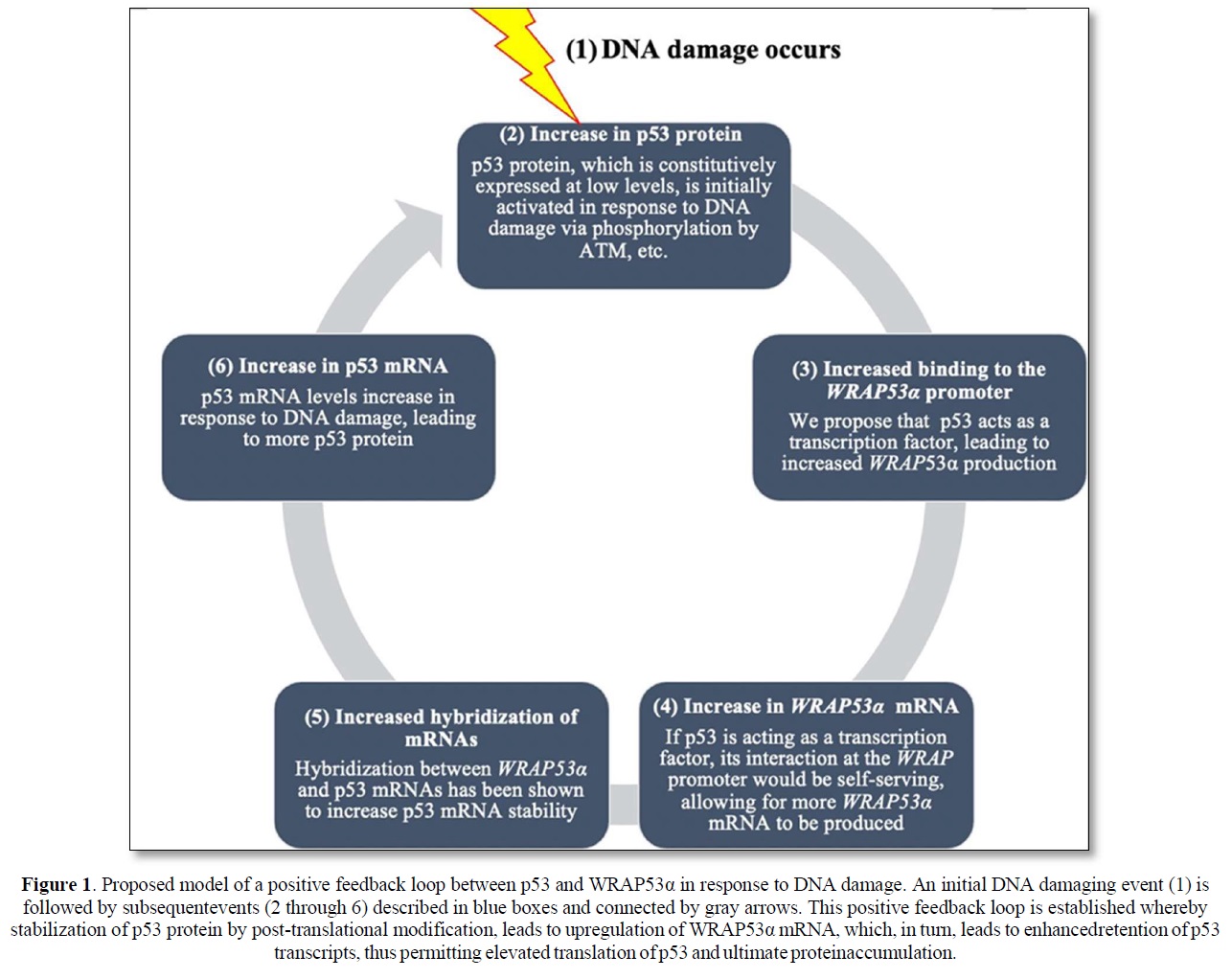
Although much remains to be validated and extended, the collective implication of these findings is the identification of what may be a positive feedback loop dependent upon WRAP53α RNA’s ability to stabilize the p53 mRNA transcript and generate more p53 protein, and p53 protein’s ability to transcriptionally stimulate WRAP53α mRNA production. The rapid increase in p53 protein levels due to initial post-translational modification would then result in binding to and activation of the WRAP promoter leading to an additional increase in p53. An illustration presenting a model of this proposed positive feedback loop is shown in Figure 1. Notably, there is precedent for such positive feedback loops wherein p53 induces its own inducer. This is demonstrated by the interaction between p53 and Wig-1, a p53 target gene protein product, which binds to an AU element in the 3’ UTR of the p53 mRNA transcript, conveying stability and protection from de-adenylation [18].
p53 has been implicated in at least seven negative feedback loops (including one with Mdm2) and at least three positive feedback loops [19]. Harris and Levine [20] predict that some feedback loops may be turned on or off in different tissue types or stages of development. Furthermore, they predicted that this propensity for feedback loops may be a convenient way for p53 to expand its cellular network, thus allowing p53 to connect simultaneously to a numerous signal transduction pathways [19].
Interestingly, mutations in the p53 protein or mutations in the WRAP promoter, could in principle, prevent the binding to the WRAP promoter from occurring which could lead to the loss of the DNA damage response. Loss of the DNA damage response, as seen with other mutations in p53, is predicted to contribute to tumorigenesis. Alternatively, specific mutations in p53 that maintain their ability to activate gene expression, may in some cases, enhance the interaction with the WRAP promoter. This could result in an accumulation of gain-of-function mutant p53 that would further contribute to tumor progression. In this respect the relationship between WRAP53 expression and rectal cancer identified by Zhang [20] may indicate a role for WRAP53 in cancer progression and deserves further investigation. In cases such as these, the ability to specifically block mutant p53 and any potential positive feedback loop involving WRAP could provide a novel approach to cancer treatment by limiting the expression of oncogenic gain-of-function mutants.
Finally, a better understanding of the regulation of expression of Wrap53 and its involvement in the cell’s response to DNA damage will add to the current knowledge of the pathways responsible for the response to DNA damage. That p53, itself regulated by Wrap53, may also participate in a positive feedback loop leading to a further increase in p53 mRNA and protein, demonstrates the dynamic and complex nature of the DNA damage response. Further studies will determine the role that various post-translational modifications in p53 have on its ability to participate in regulating Wrap53 and thus help unravel the functional nature of post-translational modifications in p53 and their role in the normal DNA damage response and how defects of alterations in this response can give rise to human cancer.
ACKNOWLEDGEMENT
This work was supported by funding from the University of South Carolina ASPIRE-I through the Vice President’s Office for Research.
CONFLICT OF INTEREST STATEMENT
There are no conflicts of interest.
REFERENCES
No Files Found




Internationally Accepted
Share Your Publication :