
-
Publish Your Research/Review Articles in our High Quality Journal for just USD $99*+Taxes( *T&C Apply)
Offer Ends On
Publish Your Research/Review Articles in our High Quality Journal for just USD $99*+Taxes( *T&C Apply)
Offer Ends On
Muhammad Daffa Hanifan, Mardhatillah Sariyanti, Besly Sinuhaji, Enny Nugraheni, Diah Ayu Aguspa Dita and Elvira Yunita*
Corresponding Author: Elvira Yunita, Department of Biochemistry and Molecular Biology, Faculty of Medicine and Health Sciences, University of Bengkulu, Bengkulu, Indonesia.
Received: February 12, 2024 ; Revised: March 02, 2024 ; Accepted: March 05, 2024 ; Available Online: March 22, 2024
Citation: Hanifan MD, Sariyanti M, Sinuhaji B, Nugraheni E, Dita DAA, et al. (2024) In Silico Analysis of Flavonoid Potential as Inhibitors of Interleukin-6 (IL-6) and Tumor Necrosis Factor-Alpha (TNF-α) in Managing Cytokine Storm. J Pharm Sci Drug Discov, 3(1): 1-10.
Copyrights: ©2024 Hanifan MD, Sariyanti M, Sinuhaji B, Nugraheni E, Dita DAA, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Views & Citations
Likes & Shares
Abstract
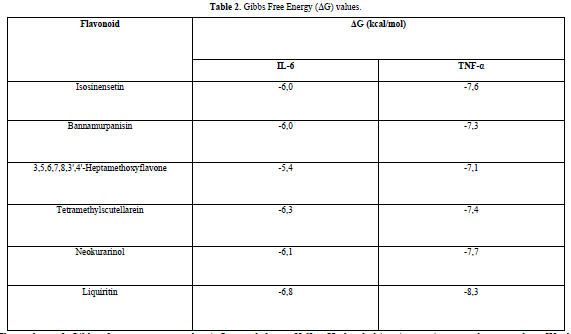
Interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α) exert the greatest influence on cytokine cascades via the nuclear factor-kappa B (NF-B) and janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathways. Through in silico analysis, the potential of flavonoids as inhibitors of IL-6 and TNF- has not been thoroughly investigated. Therefore, the purpose of this study is to investigate the insilico inhibition potential of flavonoids against IL-6 and TNF-. Through Swiss-ADME website, the Lipinski's Rule of five criteria will be applied to the test ligands. Using BIOVIA Discovery Studio and AutoDock v4.2 software, ligand and receptor preparations will be performed. AutoDock Vina software will be utilized for the mooring procedure. Observed parameters include Gibbs free energy (G) values and ligand-residue interactions that occur between the protein and ligand. Isosinensetin, bannamurpanisin, 3,5,6,7,8,3',4'-Heptamethoxyflavone, tetramethylscutellarein, neokurarinol, and liquiritin are the six flavonoids that pass the Lipinski's Rule of five criteria test when applied to 12 flavonoids. Docking results indicate that all test ligands can readily bind to both IL-6 and TNF- (G0 kcal/mol). With G values of -6.8 kcal/mol and -8.3 kcal/mol for IL-6 and TNF-, respectively, Liquiritin is predicted to have the highest potential to inhibit these proteins.
Keywords: Cytokine storm, Flavonoid, Molecular docking
INTRODUCTION
Cytokine storm can be induced by monogenic disorders, chimeric antigen receptor (CAR) T-cell therapy, cancer, and infections [1]. Cytokine storms are characterized by an increase in pro-inflammatory cytokine levels, particularly interleukin-6 (IL-6), IL-8, tumor necrosis factor-alpha (TNF-α), and interferon (IFN)-γ [2]. TNF-α and IL-6 are believed to play a significant role in the cytokine storm process [3]. TNF-α binding to its receptor activates the nuclear factor-kappa B (NF-κB) signaling pathway, inducing the production of pro-inflammatory cytokines like granulocyte macrophage-colony stimulating factor (GM-CSF), IL-8, and intercellular adhesion molecules (ICAMs), exacerbating inflammation [4]. In the cytokine storm process, IL-6 is primarily involved in the activation of janus kinase/signal transducer and activator of transcription (JAK/STAT) through two pathways, classical cis-signaling and trans-signaling Activation of these pathways leads to increased differentiation of T helper 17 (Th17) cells, cluster of differentiation 8 (CD8+) T cells, and B cells, as well as increased secretion of various inflammatory factors, including vascular endothelial growth factor (VEGF), monocyte chemotactic protein-1 (MCP-1), IL-8, and more IL-6, thereby increasing the likelihood of cytokine storms [5].
Currently, some drugs used to treat cytokine storms are monoclonal antibodies, such as tocilizumab and sarilumab as IL-6 inhibitors, and adalimumab and etanercept as TNF-α inhibitors [4]. However, monoclonal antibodies have complex manufacturing operations involving biological raw materials and stringent procedures to ensure tolerability and product quality. The production of antibody therapy more expensive to develop and produce compared to most small-molecule drugs [6]. All potential compounds with anti-inflammatory effects need to be investigated, including natural compounds, one of which is flavonoid. Flavonoids are secondary metabolites found in plants with a polyphenolic structure that exhibits various biological effects, including anti-inflammatory, antioxidant, and immunomodulatory activities [7].
Kalamansi orange (Citrus microcarpa) is one of the widely cultivated commodities in the Bengkulu Province [8]. Flavonoids are one of the groups of compounds abundant in kalamansi orange [9]. Currently, there is limited literature on the mechanism of flavonoids as anti-inflammatory agents based on their interactions with IL-6 and TNF-α. Therefore, further research is needed to investigate the potential of flavonoids in inhibiting the inflammatory process based on their interactions with IL-6 and TNF-α.
Molecular docking is a widely used method in drug discovery and development. This method is performed in silico (computational simulation) to predict the binding and affinity of a molecule with the binding site on the target receptor [10]. This can enhance the efficiency and effectiveness of research by identifying specific test components from the beginning [11]. Therefore, researchers are interested in studying the interactions between IL-6 and TNF-α with flavonoid in silico.
MATERIALS AND METHODS
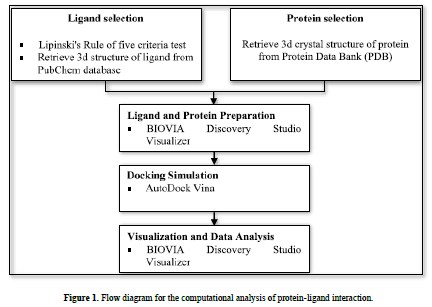
This research used a laptop with the specifications of an intel® core i7-6500U processor, 4.00 GB of Random-Access Memory (RAM), along with several software, namely AutoDock v4.2, AutoDock Vina, and BIOVIA Discovery Studio Visualizer. All software was run on the Windows 10 operating system. The materials used were 3D structures of flavonoids downloaded from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) and 3D crystal structures protein downloaded from the Protein Data Bank (PDB) (www.rcsb.org) (Figure 1).
Lipinski's Rule of Five Criteria Test
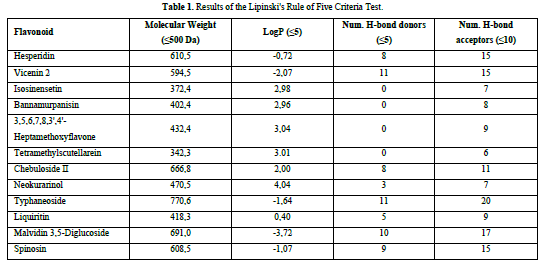
The flavonoid compounds to be used as ligands in this research will undergo the Lipinski's Rule of five criteria test. The criteria include a molecular weight ≤ 500 Da, a LogP value ≤ 5, the number of hydrogen bond donors ≤ 5, and the number of hydrogen bond acceptors ≤ 10. The Lipinski's Rule of Five criterion test can be conducted by extracting the simplified molecular input line entry system (SMILES) code of the required chemicals from the PubChem website (https://pubchem.ncbi.nlm.nih.gov/) and afterwards inputting it into the Swiss-ADME website (http://www.swissadme.ch/).
Ligand and Protein preparation
The 3D structure of the ligands is downloaded in .sdf format from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). The ligands, previously saved in .sdf format, were converted to .pdb format using BIOVIA Discovery Studio Visualizer software. Next, preparation was carried out, which involved torsion setting, as well as the addition of hydrogen atoms and gasteiger charge assignment using AutoDock v4.2 software. After the preparation, the ligand file was saved in. pdbqt format.
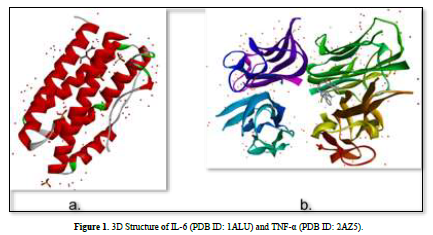
The 3D structures of the IL-6 protein (PDB ID: 1ALU) and TNF-α protein (PDB ID: 2AZ5) were gotten from the Protein Data Bank (PDB) website (www.rcsb.org) in .pdb format (Figure 2). The preparation of protein involves the separation of water molecules and native ligand using BIOVIA Discovery Studio Visualizer software. Subsequently, the protein is optimized using the AutoDockTools software to add polar hydrogen atoms and kollman charge, and then saved in. pdbqt format.
Docking Validation
Docking method validation was performed by redocking the protein with the native ligand, which was previously separated during the preparation process. The validation steps were aimed at obtaining the grid box center coordinates based on the center coordinates of the native ligand. The parameter observed during validation was the root mean square deviation (RMSD) value. The method is considered valid if the RMSD value obtained is ≤ 2˚Å, allowing the docking of test compounds with the target protein using the same dimensions and grid box coordinates. This process was conducted using AutoDock v4.2 software.
Docking Simulation
The ligands and proteins, which have been saved in pdbqt format, will undergo docking simulations using AutoDock Vina software. The simulations will be executed using the command prompt, where the command is provided as follows: "location of AutoDock Vina\vina.exe" --receptor (protein file name) pdbqt --ligand (ligand file name) pdbqt --config config.txt --out output.pdbqt. Docking simulations will be repeated ten times for each ligand to obtain the best Gibbs free energy (ΔG) values.
Data Analysis
The analysis of the research results is evaluated based on several parameters, including the values of Gibbs free energy (ΔG) obtained from the docking of each ligand to the protein receptor, the ligand-protein complexes, and the interactions between residues and ligands visualized using BIOVIA Discovery Studio Visualizer.
RESULTS AND DISCUSSION
Lipinski's Rule of Five Criteria Test
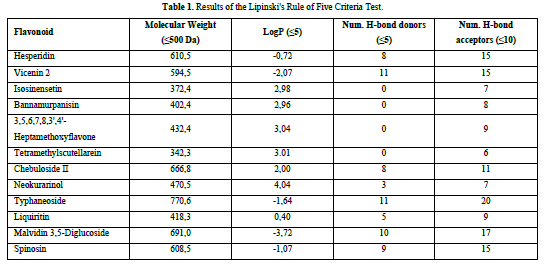
A compound is considered to meet the Lipinski's Rule of Five criteria if it fulfills three out of the four criteria. Test ligands that violate more than one criterion are not further processed in the molecular docking procedure. Based on the results of the Lipinski's Rule of Five criteria test (Table 1), it was found that isosinensetin, bannamurpanisin, 3,5,6,7,8,3',4'-Heptamethoxyflavone (HMF), tetramethylscutellarein, neokurarinol, and liquiritin have a molecular weight ≤ 500 Da, indicating that these six test ligands can pass through the cell membrane. On the other hand, the other six compounds have a molecular weight > 500 Da, predicting their inability to pass through the cell membrane [12]. All 12 test ligands have a LogP value ≤ 5, indicating their low hydrophobicity. Hydrophobicity plays a crucial role in determining drug distribution and the rate of drug metabolism in the body. A low hydrophobicity suggests that these three ligands can be easily absorbed and penetrate the lipid bilayer membrane [13]. The subsequent condition pertains to the limitation of hydrogen bond donors to a maximum of five, as well as the restriction of hydrogen bond acceptors to a maximum of ten. The compound's capacity to establish hydrogen bonds and its absorption rate are influenced by the quantity of hydrogen bond donors and acceptors. The more hydrogen bonds formed, the higher the energy required in the absorption process of the compound [14]. The results show that only isosinensetin, bannamurpanisin, HMF, tetramethylscutellarein, neokurarinol, and liquiritin meet the criteria. Based on the Lipinski's Rule of Five criteria, it can be concluded that isosinensetin, bannamurpanisin, HMF, tetramethylscutellarein, neokurarinol, and liquiritin have good solubility in the body, allowing these six compounds to proceed to the molecular docking process.
Docking Validation
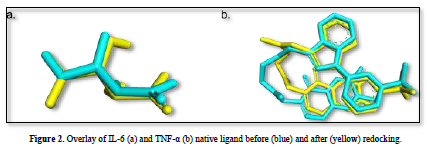
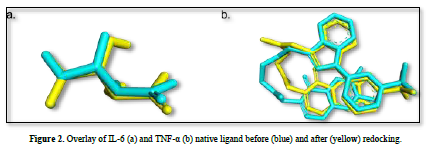
The observed parameter is the root mean square deviation (RMSD) value obtained after redocking the native ligand with the protein. The RMSD value is a parameter that indicates the extent of deviation of the native ligand before and after redocking [15]. The validation process of the IL-6 protein at grid center x = -7.677, center y = -12.743, and center z = 0.007 with dimensions 26x26x26 resulted in a root mean square deviation (RMSD) value of 1.115 Å. Meanwhile, the validation process of the TNF-α protein at grid center x = -19.163, center y = -74.451, and center z = 33.837 with dimensions 26x26x26 resulted in an RMSD value of 1.458 Å. These results can be deemed valid as the RMSD values obtained after the redocking process are ≤ 2˚Å [11]. Therefore, the same dimensions and grid box coordinates can be used for the docking process with the test ligands. Figure 3 shows the overlay of the native ligands of IL-6 and TNF-α before and after the redocking process.
Docking Simulation
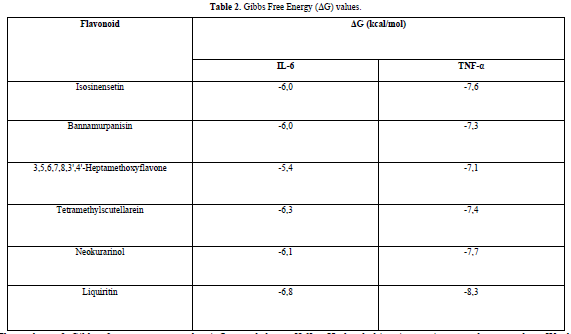
The results obtained from the docking simulations are in the form of Gibbs free energy (ΔG) values for each test ligand. The ΔG value reflects the energy involved in the docking process of the ligand and the receptor. The lower the ΔG value, the stronger the bond between the ligand and the receptor, and the more spontaneous the chemical reaction [14]. This is because when a reaction occurs, there is a release of free energy, resulting in the free energy of the product being lower compared to the free energy of the reactant [16].
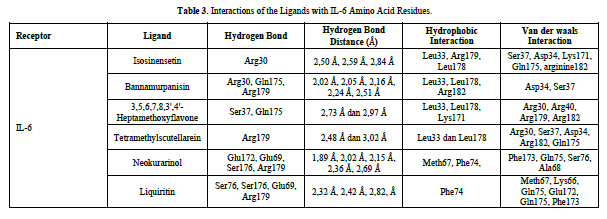
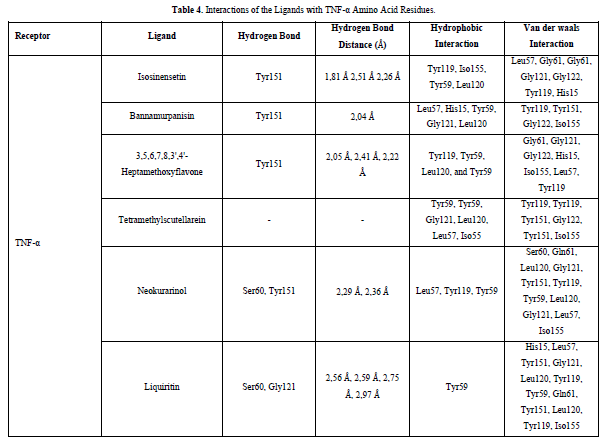
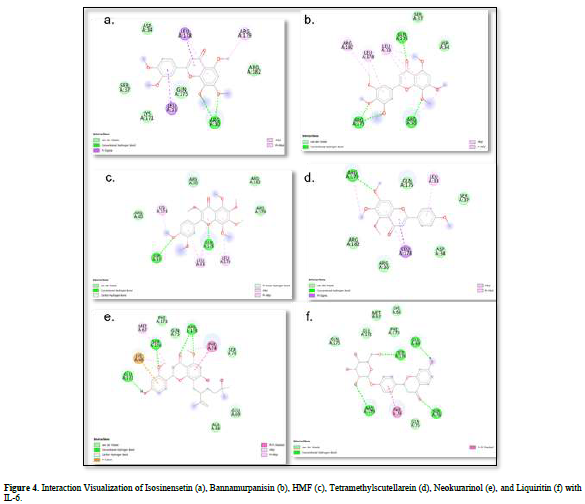
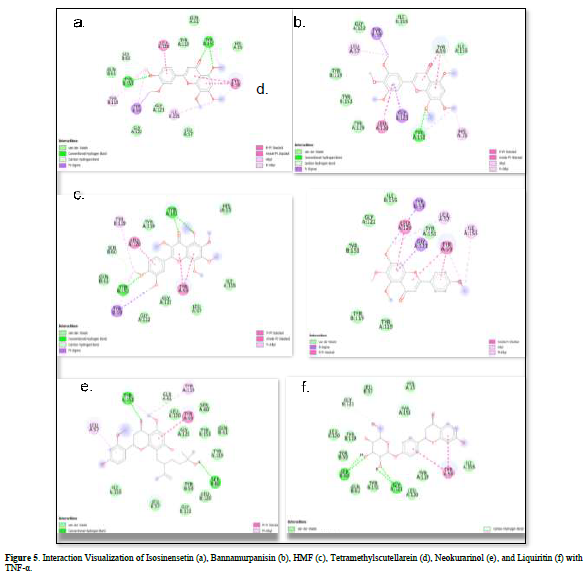
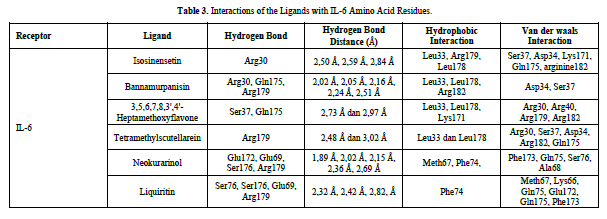
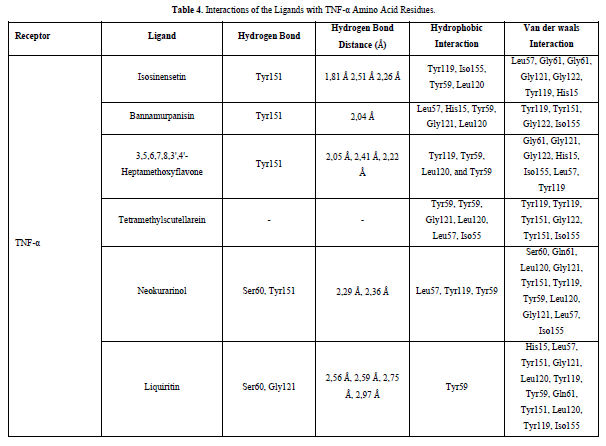
The docking results show that isosinensetin, bannamurpanisin, HMF, tetramethylscutellarein, neokurarinol, and liquiritin can spontaneously bind to both IL-6 and TNF-α. The results also indicate that liquiritin has better potential in inhibiting IL-6 and TNF-α compared to other compounds (Table 2). The interactions of the test ligands with IL-6 form bonds with amino acid residues such as Arg179, Arg182, and Gln175. This finding is in line with previous studies reported by Azzahra (2021) (2021) dand Wang (2019) (2019) [17,18] which state that the 3D crystal structure of IL-6 with PDB ID 1ALU has active sites at amino acid residues Arg179, Arg182, and Gln175. On the other hand, the interactions of the test ligands with TNF-α form bonds with amino acid residues Leu57, Tyr59, Ser60, Gln61, Tyr119, Leu120, Gly121, Gly122, and Tyr151. This finding is consistent with research conducted by Zia (2020) and Yende (2021) [19,20] indicating that the 3D crystal structure of TNF-α with PDB ID 2AZ5 contains an active site on the amino acid residues Leu57, Tyr59, Ser60, Gln61, Tyr119, Leu120, Gly121, Gly122, and Tyr151. The findings of this study suggest that isosinensetin, bannamurpanisin, and HMF have the potential as candidate inhibitors of IL-6 and TNF-α.
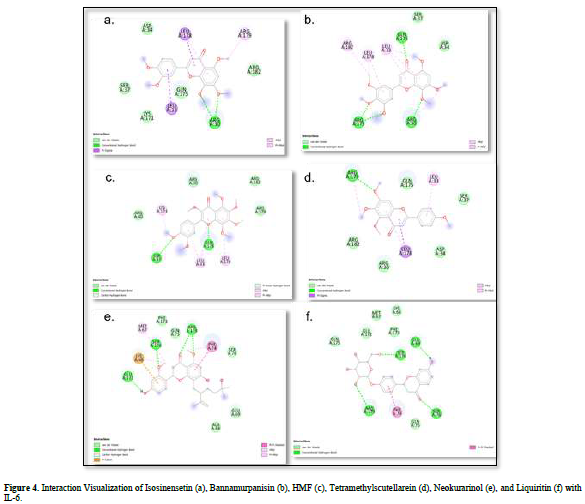
The value of Gibbs free energy can be influenced by residue-ligand interactions, such as hydrogen bonds, van der Waals interactions, and hydrophobic interactions. The more interactions that occur, the greater the ΔG value [21]. Hydrogen bonds are a type of bond that occurs between hydrogen atoms and electronegative atoms such as fluorine, nitrogen, and oxygen [22]. Hydrogen bonds are easier to form and can create stronger bonds with active sites [23]. The strength of hydrogen bonds can be influenced by the length of the bond; longer hydrogen bonds result in weaker bond strength and therefore a weaker ΔG value [24]. Hydrophobic interactions occur due to the attractive forces between hydrophobic (non-polar) molecules [25]. On the other hand, van der Waals bonds are formed due to the temporary change in dipole moment arising from the short-term electron orbital shift to one side of an atom or molecule [26]. Hydrophobic interactions and van der Waals interactions play a role in stabilizing ligand-receptor interactions, and the presence of both interactions can affect the ΔG value, even if they have relatively weak affinities [27].
Based on the residue-ligand interactions that occur between the test ligands and IL-6 (Figure 4 & Table 3), although the number and length of hydrogen bonds formed are not greater, liquiritin produces a lower ΔG value compared to other compounds. This is because more amino acid residues are involved in the interactions with liquiritin, and the more residues involved in the interaction with the ligand, the stronger the bond formed between the ligand and the receptor [28]. Additionally, liquiritin also shows more van der Waals interactions, which allow it to form more bonds compared to other compounds.
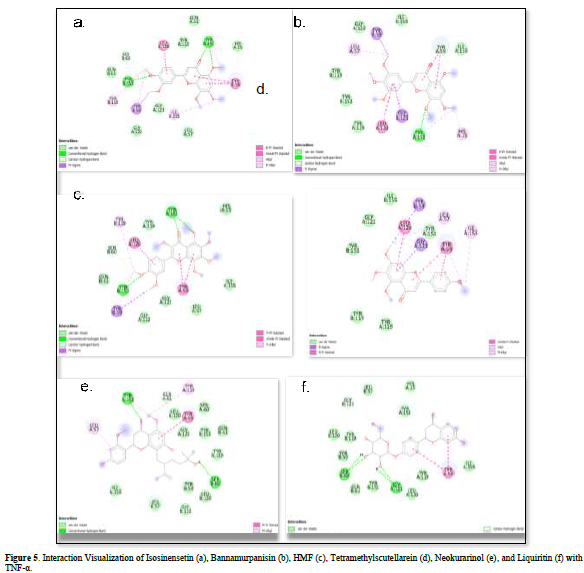
Referring to the interactions between ligand residues and TNF-α (Figure 5 & Table 4), liquiritin forms more hydrogen bonds and involves more amino acid residues in van der Waals interactions compared to other compounds, resulting in a lower ΔG value. The hydrophobic interactions that occur between bannamurpanisin and Tetramethylscutellarein with TNF-α tend to have a greater influence on the ΔG value compared to hydrogen bonding. This can be observed from the lower ΔG values of bannamurpanisin and Tetramethylscutellarein compared to HMF. Venugopal [29] stated that the more hydrophobic interactions formed, the more stable the ligand-receptor complex, leading to lower ΔG values. This stability is due to hydrophobic interactions enabling the protein to reduce the surface area and minimize unwanted interactions with water molecules [30]. However, it should be understood that there are factors influencing ΔG value that cannot be observed in molecular docking. To gain a more comprehensive understanding of the structure-function relationship of proteins and protein-ligand complexes, as well as the ΔG value, it is important to conduct further in-depth analysis and verification with other methods, such as molecular dynamics [31].
Effectiveness of Flavonoid Compounds from Molecular Docking in Inhibiting IL-6 and TNF-α
The results of this study showed that all six test ligands were able to bind to the active sites of IL-6 and TNF-α, preventing these cytokines from binding to their receptors and inhibiting the cascade of signaling that triggers massive inflammation. Interleukin-6 (IL-6) is a multifunctional cytokine that is synthesized in response to either tissue damage or infection. During cytokine storms, high levels of IL-6 can lead to serious health issues. IL-6 activities in classical cis-signaling and trans-signaling pathways induce Janus Kinase (JAK) phosphorylation, subsequently activating signal transducer and activator of transcription (STAT), especially STAT [32]. This leads to an increase in the secretion of inflammatory factors contributing to systemic inflammation, which can cause organ damage, organ dysfunction, and severe symptoms such as high fever, hypotension, respiratory distress, and other organ function impairments [33]. TNF-α has the ability to increase blood vessel permeability during viral infections and plays a crucial role in several autoimmune diseases and chronic inflammation [34]. However, excessive TNF-α production, as observed during cytokine storms, can have negative effects on the body [35]. TNF-α is a major inducer of NF-κB, responsible for the expression of multiple inflammatory genes in the cell nucleus. The stimulation of other pro-inflammatory cytokines, such as TNF-α, is induced by this process, resulting in a robust and persistent inflammatory reaction [36]. Inhibiting the binding of IL-6 and TNF-α to their receptors. The prospective technique of limiting the binding of IL-6 and TNF-α to their receptors has been identified as a means to inhibit excessive inflammation and mitigate adverse effects on the body. This approach has promise for the advancement of effective anti-inflammatory medicines [37].
Flavonoid known as compound in broad natural product. One of that is from Kalamansi orange from Bengkulu Province, Indonesia [9]. This study suggests that flavonoid from Kalamansi oranges was potential candidate for inhibiting pro-inflammation’s cytokines through in silico study. Flavonoid could inhibit activity of TNF-α and IL-6 through inhibits nitric oxide synthase production in nf-kB pathway. Thus, the effect of this mechanism could inhibit the activity of TNF-α and IL-6 [38]. However, in real time this study still has limitations. The interaction of the compound with IL-6 and THF-alfa proteins has not been analyzed using molecular dynamic tests. Thus, it cannot be predicted dynamically the binding between the compound and the cytokine molecule. Apart from that, due to limitations in in silico studies, it is still necessary to develop further analysis at the in vitro and in vivo stages
CONCLUSION
Liquiritin is predicted to be the most potent compound in inhibiting IL-6 and TNF-α with Gibbs free energy (ΔG) values -6.8 kcal/mol and -8.3 kcal/mol, respectively. It is necessary to conduct tests using other in silico methods such as toxicity testing and molecular dynamics analysis before performing in vivo and in vitro tests. Toxicity testing is required to gain an understanding of the potential of a compound to cause damage to cells or organs when exposed to an organism. Meanwhile, molecular dynamics analysis is needed to ensure the stability and strength of interactions between ligands and receptors after the docking process.
CONFLICTS OF INTEREST
The authors declare that there is no conflict of interest regarding the publication of this paper.
ACKNOWLEDGMENT
The authors express their gratitude to the Faculty of Medicine and Health Sciences of the University of Bengaluru for their cooperation in facilitating the execution of this research.
No Files Found




Internationally Accepted
Share Your Publication :