
-
Publish Your Research/Review Articles in our High Quality Journal for just USD $99*+Taxes( *T&C Apply)
Offer Ends On
Publish Your Research/Review Articles in our High Quality Journal for just USD $99*+Taxes( *T&C Apply)
Offer Ends On
Michel Goldberg*
Corresponding Author: Michel Goldberg, Department of Oral Biology, Faculty of Fundamental and Biomedical Sciences, Paris Cité University, France.
Received: April 28, 2023 ; Revised: May 03, 2023 ; Accepted: May 06, 2023 ; Available Online: May 31, 2023
Citation: Goldberg M. (2023) Rares Human Genetic Disorders. J Oral Health Dent Res, 3(1): 1-4.
Copyrights: ©2023 Goldberg M. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Views & Citations
Likes & Shares
Single and multifactorial genes, chromosome abnormalities and mitochondrial genetic inheritance disorders contribute to the 8000 to 10,000 genetic inheritance disorders. Trisomy 21 (Down syndrome), Turner syndrome (X0), triple X syndrome (XXX), Klinefelter syndrome, constitute a few examples of gene anomaly reviewed in this article. Altogether, these anomalies include many others diseases such as Patau syndrome (trisomy 13), Edwards syndrome (trisomy 18), cri du chat syndrome, cystic fibrosis, fragile X syndrome, Prader-Willi syndrome, Huntington’s disease, muscular dystrophy (Duchenne) and mitochondrial genetic inheritance disorders that play a role in the etiology of genetic disorders. These pathologies are resulting from gene mutations contributing to clarify the causes of some genetic pathologies. They further clarify the origin of some human disorders implicated in genetic alterations and shed lights on the implication of some gene’s disorders in human syndromes.
Keywords: Gene single mutation, Multifactorial genes, Down syndrome, Turner syndrome, Klinefelter syndrome, Chromosomal abnormalities
INTRODUCTION
Health problem are caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosomal abnormality. More than 300 million people are living with rare diseases having a genetic basis. These genetic disorders are either autosomal recessive or dominant inheritance. Some genetic disorders are classified by a mutation X-linked., and very few disorders are inherited on the Y chromosome or mitochondrial DNA.
Rares diseases refer to 1/2000 people. They are classified as genetic disorders.
Four disorders have been identified that are resulting from:
There are 8000 to 10 000 genetic diseases that are including chromosomic abnormalities, monogenic (mutation of one single gene (responsible for about 7000 pathologies) and polygenic mutations due to the interaction of different modifications).
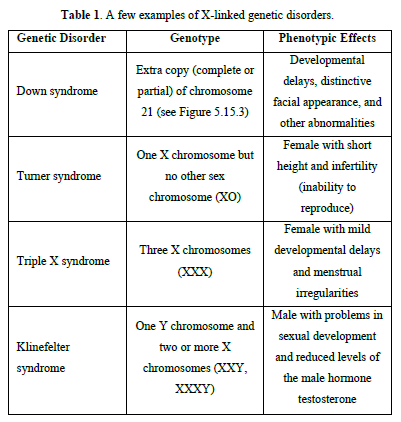
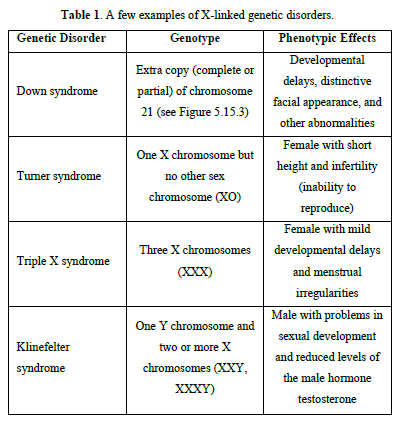
A few examples of X-linked genetic disorders are listed in Table 1.
Trisomy 21: An extracopy of chromosome 21. (e.g. Trisomy 21 [1-3], hemophilia that concern essentially boys, gene being located on chromosome X, Duchenne myopathy or neuromuscular sickness, drepanocytes anomaly of hemoglobin present in red blood cells).
Mitochondrial: Leber’s hereditary optic neuropathy.
Other genetic diseases affecting patients:
Sickness of Charcot-Marie-Tooth, Duchenne dystrophy muscular, hyper cholesterolemia familial, Gaucher’s disease, Hemophilia, Huntington’s disease, porphyria, thalassemia, Syndrome of Turner, Tay-Sachs disease, Wilson disease, Parkinson disease, Down syndrome, autism, sickle cell anemia.
In addition to monogenic and polygenic mutations, chromosomal abnormalities, and genetic diseases have been identified:
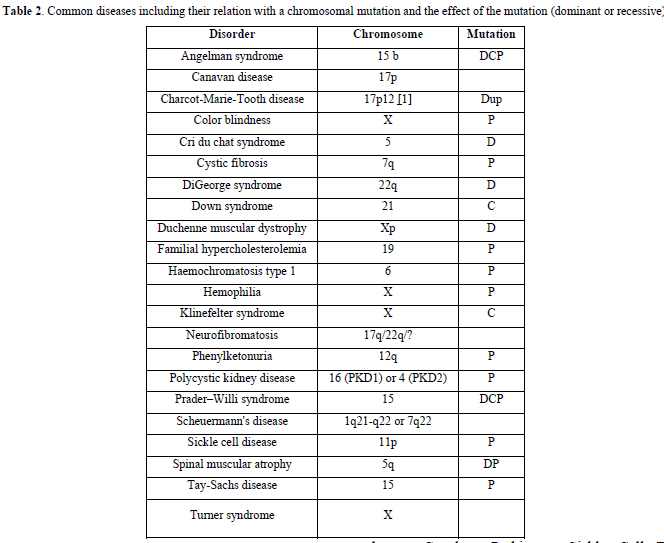
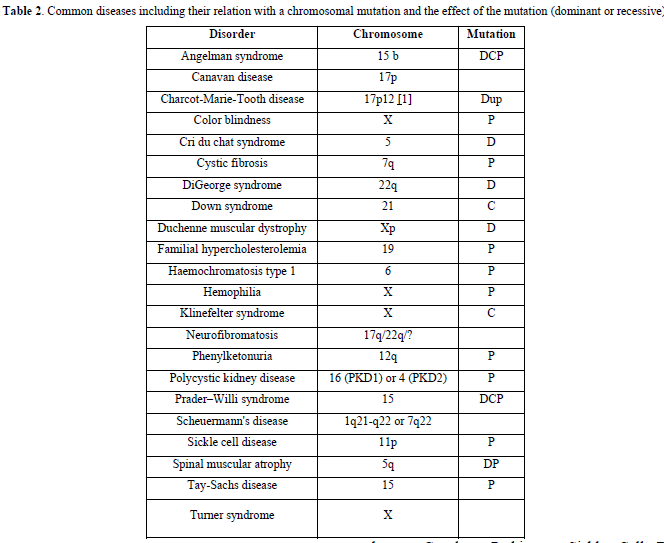
Genetic disorders are also listed:
Achondroplasia, antiphospholipid syndrome, autism, Charcot-Marie-Tooth disease, Crohn ‘s disease, Down syndrome, Duchenne muscular dystrophy, fragile X syndrome, Gaucher, Parkinson, Sickle Cell Disease, Thalassemia, and Turner syndrome.
A few genetic disorders
Darier sickness or dyskeratose follicular. (2 for 100 000 individuals). This disease cannot be cured. Gene mutation of chromosome 12 gene, called ATP2A. coding for a defective gene (75% patients are sick before 20 years).
Down syndrome
Trisomy 18 and trisomy 13. One patient for 800 and 1/1000 live born infants have the syndrome [1-3].
Signs of the sickness: heart defect, low thyroid symptoms, eye problems (cataracts), a flattened face, short (flat) nose bridge, small ears, short neck, decreased muscle tone, heart and digestive system defects, small hands and feet, and developmental delays.
There is a total 47chromosomes, instead of 46 (46 Chromosomes +1).
Down syndrome displays an autosomal dominant or recessive pattern.
Down syndrome
Cystic Fibrosis is a genetic disorder caused by a mutation of the CFTR (cystic fibrosis transconductance regulator) gene that controls the movement of salt and water in and out of the cells.
CF is not curable, but may be managed. Although cystic fibrosis is progressive and requires daily care, children and people with CF can usually attend school and work. They often have a better quality of life than people with CF had in previous decades. Improvements in screening and treatments mean that people with CF may live into their mid-to-late 30s or 40s, and some are living into their 50s.
Cystic fibrosis is a monogenic disease considered to affect at least 100 000 people worldwide. Mutations in CFTR, lead to impaired clearance. Classical cystic fibrosis has been characterized. This autosomal recessive disease is diagnosed in many regions following newborn screening, whereas in other regions, diagnosis is based on a group of CFTR mutations. This disease is less easily diagnosed. Management strategies have gradually improved life expectancy for people with cystic fibrosis.
Fragile X syndrome [4,5]
Sometime associated with autism (1/1500 males and 1/2500 females). Familial combined hyperlipidemia and familial hypercholesterolemia are inherited disorders that result in an increase in blood lipids and cholesterol levels. Treatment includes lifestyle and dietary changes. Diagnosis requires genetic testing to determine the number of CGG repeats in the FMR1 gene. Normally, there are between 5 and 40 repeats. Fragile X syndrome is the most "translated" human neurodevelopmental disorder under study. Evidence from mouse models shows that mGluR5 antagonists (blockers) can rescue dendritic spine abnormalities and seizures, may show promise in the treatment of FXS. Two new drugs, mavoglurant and dipraglurant, as well as the repurposed drug fenobam are currently undergoing human trials for the treatment of FXS.
Huntington’s disease is an inherited disease which causes certain nerve cells in the brain and central nervous system to degenerate. Usually develops when a person is in their 30’s and 40’s. There is also an early-onset form of the disease, which begins in childhood.
loss of these nerve cells causes symptoms such as behavior changes, unusual, snake-like movements (chorea), uncontrolled movement, difficulty walking, loss of memory, altered speech and cognitive functions, and difficulty in swallowing.
Muscular dystrophy (Duchenne): Associated with heart and respiratory problems. The muscle weakness makes it progressively more difficult to walk and get around. That weakness gets progressively worse - by the age of 12, most kids will need a wheelchair. Boys are more likely to inherit this disorder. Becker muscular atrophy require a wheelchair by the age of 25 to 30.
Symptoms include fatigue, possible intellectual disability, and muscle weakness, beginning in the legs and then spreading to the upper body.
Thalassemia: Beta-thalassemia is most common, alpha thalassemia is less common. Hemoglobin is not properly synthetized by red blood cells. Patients may also have poor appetite, darkened urine and jaundice (a yellowish discoloration of the skin or the white parts of the eyes - jaundice is a signal for liver dysfunction).
Wilson disease
Wilson disease is a rare genetic condition that causes a person's body to store too much of the mineral copper. Over time, the extra copper can lead to organ damage that may cause death.
Affect 1/30 000 people, they store too much copper. Copper storage disease induce hepato-lenticular degeneration.
Symptoms include Jaudice, bruiting clumbiness, trembling, difficulty walking, problems with speech, with scholar work, depression, anxiety, green-to-brownish rings around the iris of the eye, Anemia with low level of red blood cells, white blood cells (anemia), and low levels of platelets.
Treatment, Reduce intake of copper-containing food, excess store of copper in the brain, and in the corneas of the eyes. ATP7A and ATP7B, two homologous copper transporting P1B-type ATPases play crucial roles in cellular copper homeostasis. Menkes disease is characterized by systemic copper deficit. Hepatic copper overload and toxicity give rise to Wilson disease. The gene is located on chromosome 13, coding for ATPase 78. It is an autosomic recessive illness.
2-9-Coffin-Siris syndrome (and Nicolaides-Baraitser syndrome)
Developmental delays, feeding difficulties and issues with vision and hearing. Professionals refer to Coffin-Siris syndrome, and short stature-onychodysplasia.
Affect multiple bodily systems including the skeletal system, movement and dexterity, vision, hearing, speech. The cause of the disease is the mutation of one or more gene(s):
Coffin-Siris syndrome includes: coarse facial features, a thick lower lip, a wide mouth, increased bodily hair, feeding difficulties. 1/1000 000 and: heart abnormalities, urogenital abnormalities, recurrent infections, delayed skeletal maturation and growth, underdevelopment of the fifth toe or finger or the fifth toenails or fingernails, loose joints, low muscle tone, sparse scalp hair, hearing difficulties, strabismus, when the eyes point in different directions, seizures, and behavioral abnormalities including Intellectual disability deficiency, and de Novo mutation in the SMARCA2 gene.
Tay-Sachs syndrome is due to a missing enzyme called hexoaminidase A, located on chromosome 15 q23- q 24. Patients develop progressive severe mental and developmental retardation/destruction of the nervous system and brain.
Start at 4 to 8 months old child. The death is occurring by the age of 3 to 5 years whereas juvenile form age 15.
1 Patient for 3600 is involving mostly Ashkenase jews. A red spot appears on the retina of the eye. After the first 6 months post-natal, the child becomes paralytic. Death occurs before 4 years. The target is a subgroup of lipids.
Niemann-Pick disease (Type A)
Fatty material (or lipids) starts to accumulate in liver, spleen, lung and brain. Lipid storage disorders called sphingolipidoses are members of the family of lipidosis. Accumulation of lipids occurs in the spleen, liver, and in the central nervous system. Lipids are cleaved as members of the phosphomyelin components. Type A occurs in enfants.
Klinefelter syndrome
Concern 1 man for 600 newborns. The total number of chromosomes is 47 identified as 47, XXY (instead of 46, XY - with a normal distribution in male. Children display. Infertility, and small poorly functioning testicles. The extra X chromosome is a characteristic of the KS. Enlarged breasts may be removed by surgery. Hormonotherapy may ameliorate the disease.
CONCLUSION
Genetic diseases are numerous and clarify different targets. Altogether, they constitute the rare diseases family, and they take place in diverse pathologies, some of them being lethal while others lead to deep handicap. Most of them have no treatment. Specific drugs provide some ameliorations but most of the time genetic therapies leads to death. In this review we summarize the origin and phenotype of diseases and syndromes [6-8]. Identification of mutation provides genetic identification of the genes but don’t allow to cure the disease unless gene therapy is expected. However, such treatments are still not available.

No Files Found




Internationally Accepted
Share Your Publication :